
SugarQBits is a collection of software tools implemented in the cross-platform programming language Perl. The individual SugarQBits are stand-alone scripts that can be freely arranged to process any MS/MS data in the .mgf file format.
MS/MS data conversion: We routinely extract and convert MS/MS data (usually the Thermo .raw file format) using either msconvert (version 3.0.22133-5eed1a6) or PEAKS X Pro Studio 10.6 (build 20201221). We use geenric MSConvert settings (i.e. “vendor”-specific peak-picking and "remove zero-samples"). This works well for nice sprectra. PEAKS allows for advanced charge-deconvolution and deisotoping of MS/MS data, as well as additional raw-data refinement (i.e. precursor mass and charge-state re-evalutation; we do not use the "merge DDA scans"option). We prefer PEAKS processed MS/MS data as input
.
SugarQBits_DD.pl
SugarQBits_DD is a simple charge-deconvolution and deisotoping tool for MS/MS data. Very simple! We recommend this tool only if no other decon/deiso algorithm available (e.g. PEAKS). The algorithm lives off the mass-precision provided by modern instruments: the script accepts .mgf files and calculates the distances between peak pairs. If neighbouring peaks are found at a distance of 0.5 or 0.33 amu (+/- a user-defined precision value; in ppm), the charge state of these ions is considered as 2+ or 3+, respectively, and 1+ masses are calculated accordingly. Charge states > 3+ are currently not considered. Peaks of charge-state 2+ and 3+ are removed from the spectrum, and replaced by their corresponding 1+ masses with their original peak. The deisotoping algorithm sums the intensity values of all peaks that comprise the respective isotope-envelope, retains only the mono-isotopic peak and assigns the summed intensity values as intensity value to the mono-isotopic peak (see example below). The charge-decovoluted and deisotoped spectra are put out in a new file ("_DD.mgf").
Original spectrum:
SugarQBits_DD is a simple charge-deconvolution and deisotoping tool for MS/MS data. Very simple! We recommend this tool only if no other decon/deiso algorithm available (e.g. PEAKS). The algorithm lives off the mass-precision provided by modern instruments: the script accepts .mgf files and calculates the distances between peak pairs. If neighbouring peaks are found at a distance of 0.5 or 0.33 amu (+/- a user-defined precision value; in ppm), the charge state of these ions is considered as 2+ or 3+, respectively, and 1+ masses are calculated accordingly. Charge states > 3+ are currently not considered. Peaks of charge-state 2+ and 3+ are removed from the spectrum, and replaced by their corresponding 1+ masses with their original peak. The deisotoping algorithm sums the intensity values of all peaks that comprise the respective isotope-envelope, retains only the mono-isotopic peak and assigns the summed intensity values as intensity value to the mono-isotopic peak (see example below). The charge-decovoluted and deisotoped spectra are put out in a new file ("_DD.mgf").
Original spectrum:
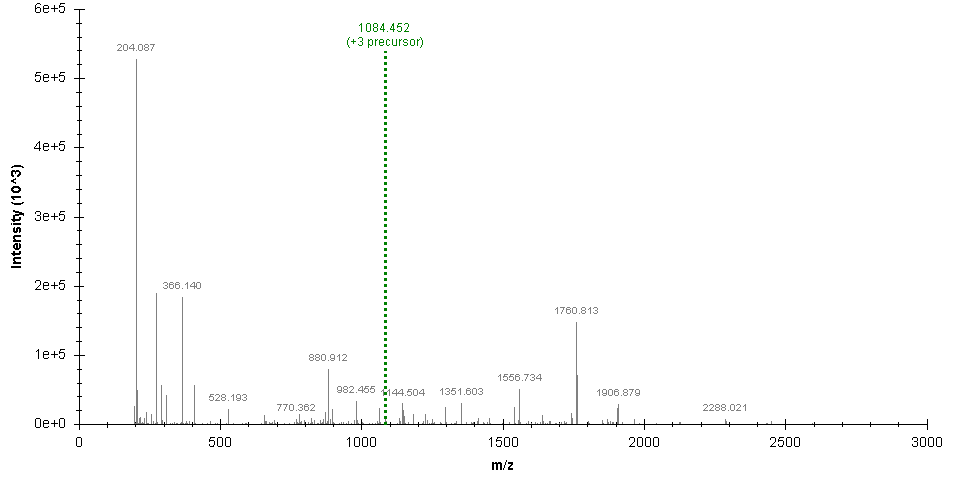
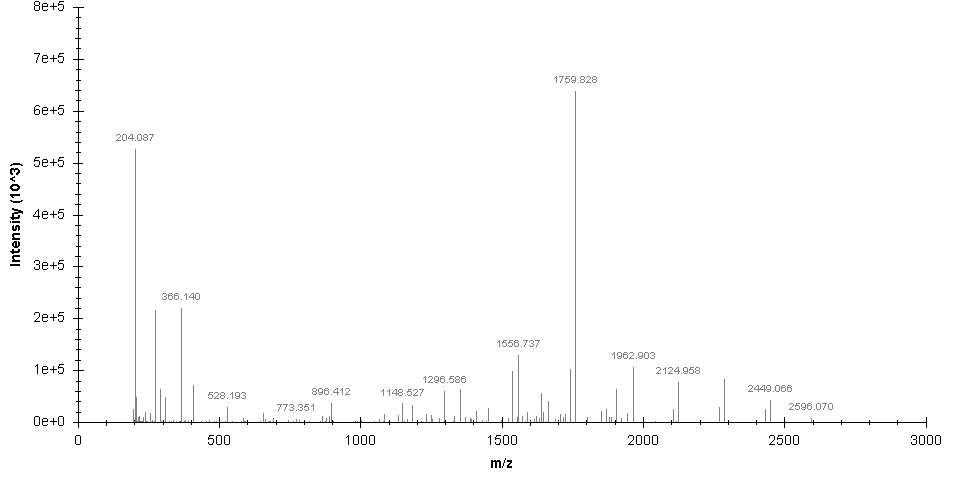
SugarQBits_DD processed spectrum:
SugarQBits_RepX.pl
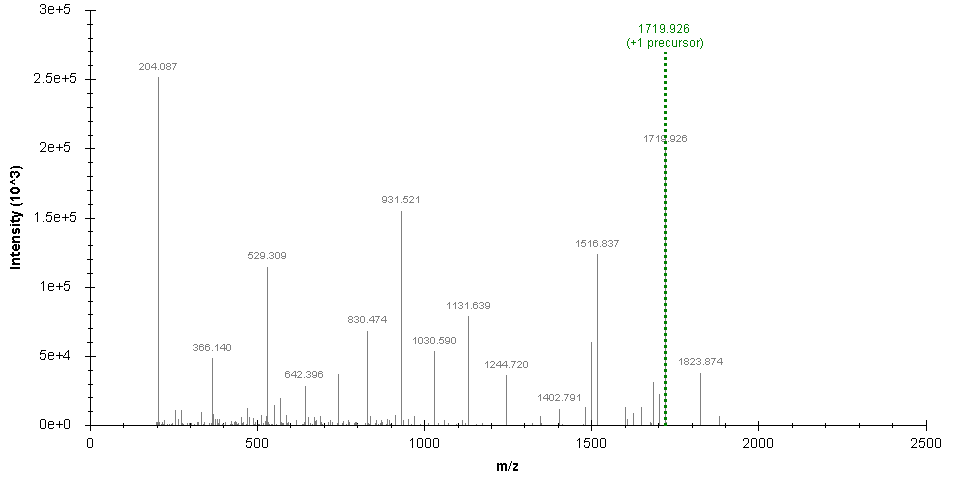
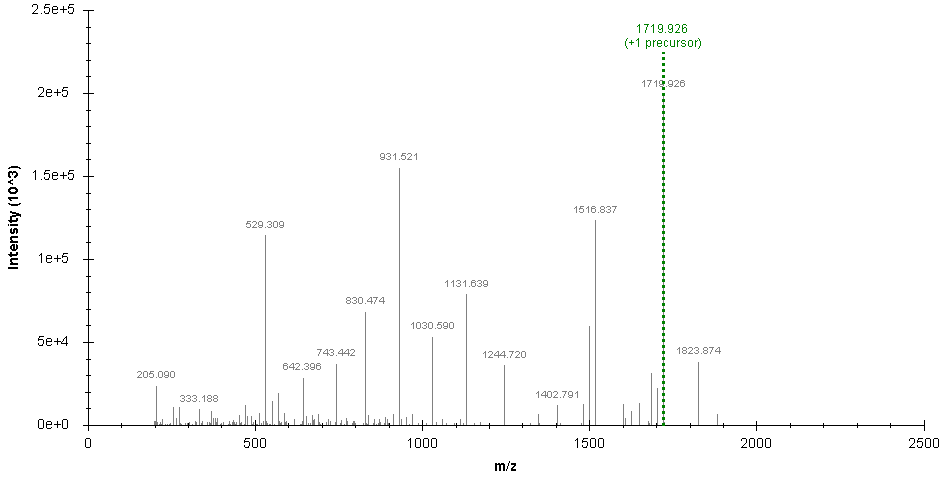
SugarQBits_RepX extracts intensity values of user-defined m/z value (+/- a user-defined precision values, in amu or ppm). The script copies the respecive intensity values into a .csv file ("_RepIntensities.csv"), and removes them from a "cleaned" copy of the input .mgf-file ("_RepX.mgf"). This script can e.g. be used to extract intensity values of TMT-reporter ions or to simply remove dominant glycan-derived oxonium ion signals (see example below), prior to peptide identification.
Original spectrum:
SugarQBits_RepX processed spectrum (removing signals at 204.08 and 366.1 m/z):
SugarQBits_Kassonade.pl
SugarQBits_Kassonade is the central SugarQBit in glycoproteomics workflows. Similar to SugarQb, the script accepts .mgf files, tries to identify Y1 (i.e. peptide + HexNAc) ions and re-adjusts the spectrum precursor mass information (i.e. PEPMASS in .mgf format) accordingly. SugarQBits_Kassonade works as an "open" glyco-search tool, as it does not requires the use of a user-defined glycan mass list.
Original spectrum:
SugarQBits_Kassonade (and SugarQBits_RepX) processed spectrum
(N.B. change of PEPMASS to m/z of automatically detected Y1+ ion):
Annotated spectrum (SearchGUI; identification by Comet):
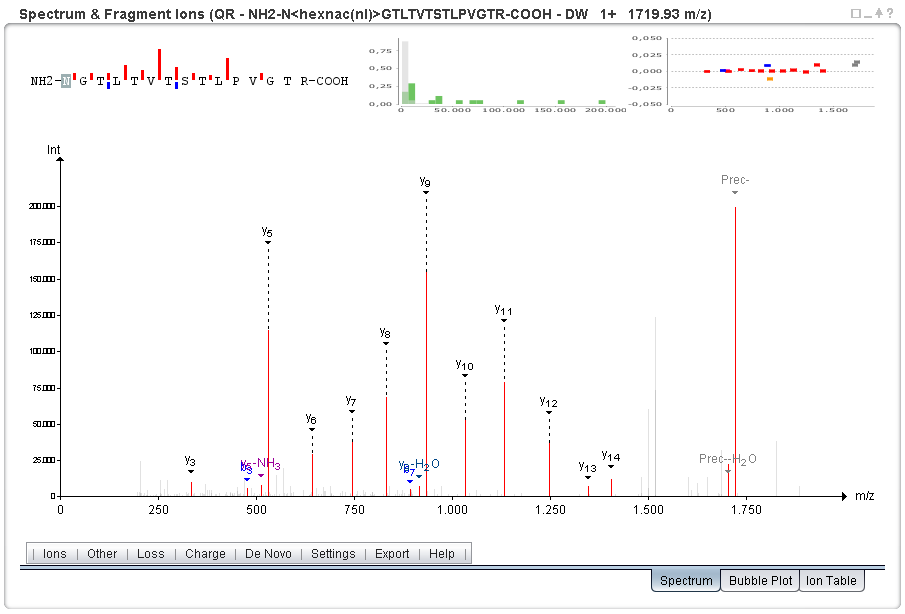
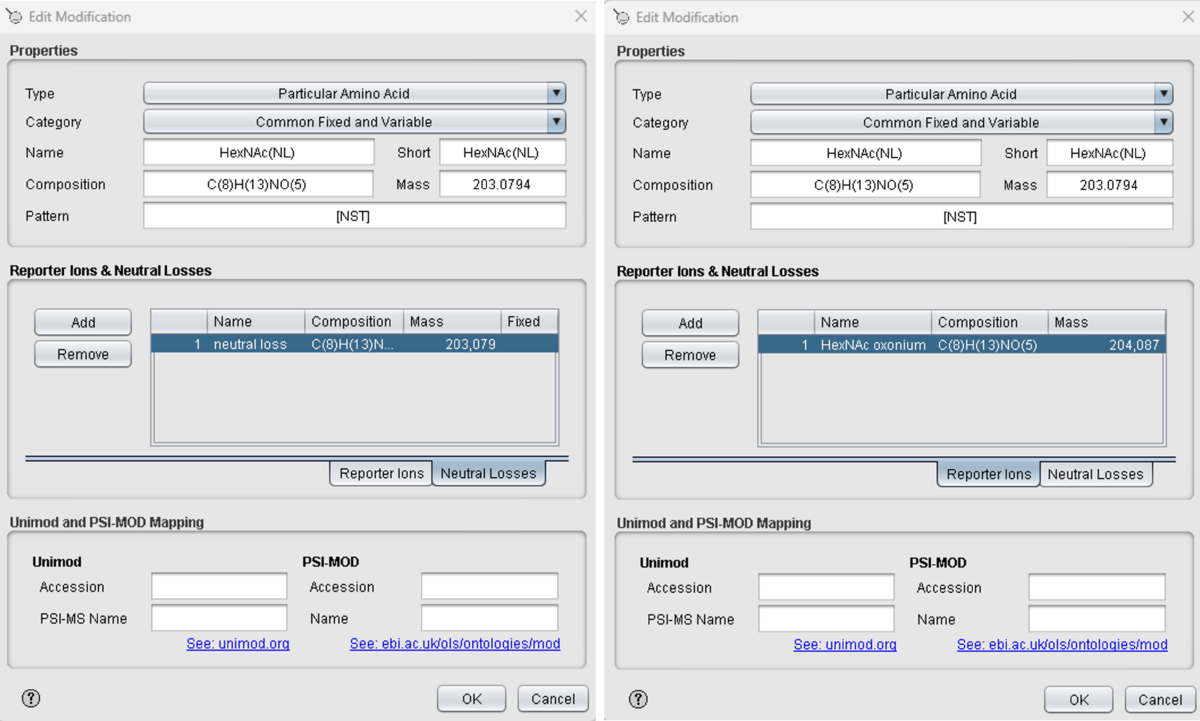
For the automated identification of glycopeptides using generic MS/MS Search Engines (e.g. MASCOT, MS-GF+, Comet, MS Amanda, OMSSA) we use the SearchGUI environment, using a custom-definition of the built-in HexNAc modifciation (see below).
SugarQBits_Kassonade.pl is a revamped version of the original SugarQb, which was developed as a collection of software tools ("nodes") to the commercial Proteome Discoverer environment (Thermo) and did not allow for an "open" search approach.
The original SugarQb nodes (and their portations to PD 2.1 and 2.3) are still freely available to all researchers at "PD Nodes" of the Protein Chemistry Facility IMP/IMBA/GMI at the Vienna Biocenter, Austria. For further information on the SugarQb algorithms and their use within PD, please refer to:
- Stadlmann J., Taubenschmid J., et al. Comparative glycoproteomics of stem cells identifies new players in ricin toxicity, Nature (2017).
- Stadlmann J., et al. Analysis of PNGase F-Resistant N-Glycopeptides Using SugarQb for Proteome Discoverer 2.1 Reveals Cryptic Substrate Specificities, Proteomics (2018).
MS2Oxoplot.pl
MS2Oxoplot is an abbreviated version of SugarQBits_RepX, merely extracting the intensity values of a list of user-defined m/z value (+/- a user-defined precision value, in amu or ppm) into a .csv file ("_RepIntensities.csv"). It does not generate a reporter ion "cleaned" .mgf-file version, but computes base-peak intensity and total ion count values for every MS/MS scan (recently used for the calculation of spectrum-specific eSNOG values).